

Pulmonary Hypertension
Our vision is to transform pulmonary hypertension (PH) into a long-term, manageable condition, so that patients can live a normal life.
Partner with Us
We are committed to partnering with those who share our vision to transform pulmonary hypertension into a long-term manageable condition.
Learn MoreLeadership
We work together to leverage scientific discovery and build on our knowledge to continue to support patients around the world.
Learn morePulmonary Hypertension
Pulmonary Hypertension
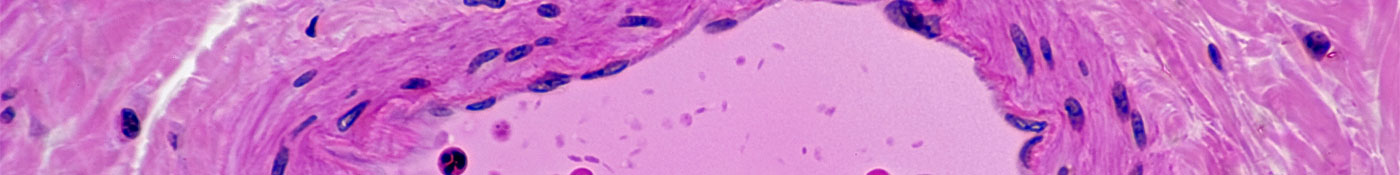
Normal Artery in Cross Section (Magnification x100) H&E stain